
Technical Note
Trimming andFiltering
The Ion Torrent system applies some quality checks to sequence reads before writing them out. Reads are tested to see if they are possibly the result of mixed DNA templates on an Ion Sphere Particle (ISP) in a given well, or if they are generally of low signal quality. The 3' ends of reads are scanned for matches to the adapter sequence and for regions of trailing low quality. Only the high-quality portions of reads passing through these checks are written out to the unmapped BAM file.
Introduction
When processing data from an Ion Personal Genome Machine (PGM ) or Ion Proton instrument, the identification and removal of low-quality or uncertain base calls is an important consideration for delivering accurate results. In the Torrent Suite Software analysis software, low-quality bases are removed from the output by:
- Trimming low-quality 3' ends of reads.
- Filtering out entire reads.
The logic by which reads are filtered and trimmed is the same for Ion PGM and Ion Proton instruments, though the two instruments differ in terms of where the logic is applied. On the Ion PGM instrument, all of the data processing occurs on the Torrent Server, as the Ion PGM instrument itself has very limited compute capabilities. The Ion Proton instrument, on the other hand, has significant compute power on-board, enabling some of the read filtering to occur during signal processing on the instrument itself.
Trimming
The goal of read trimming is the removal of undesired base calls at the 3' end of a read. Such undesired base calls come from:
- High-quality base calls on reads that have read all the way through the library insert into the 3' adapter that binds the library templates to the ISPs.
- Low-quality base calls at the 3' end of the read.
Two filters are applied to trim reads, targeted at the two categories of undesirable 3' bases above. Each filter is applied separately and the read length is taken as the shortest of the two. If the resulting trimmed length is shorter than the minimum read length (eight bases), the read is filtered out entirely.Otherwise the read is written to the unmapped BAM file.
Removal of the adapter sequence
Trailing adapter sequence is trimmed out by searching the read for candidate matches to the known adapter sequence in flow-space. If a read extends into the adapter, the 3' end of the flow signals exhibits a pattern that is characteristic of the adapter sequence.
Adapter trimming is accomplished by testing each candidate position in flow space to see how well the flow signals match the pattern expected for the adapter. The test computes the Euclidean distance between the flow signals at the tested position and the predicted flow signals for the adapter. If this distance falls below a fixed threshold, the corresponding position (translated from flow-space back to read-space) is recorded as the adapter trim location; otherwise, the read is considered not to have a match to the adapter.The default threshold used is a Euclidean distance of 16.
The 3' adapter sequence can be selected from a predefined list either during planning (Plan New Run / Kits / Library Kit Type Details / Forward 3' Adapter) or on Edit Run page. The predefined list contains adapter sequences corresponding to standard Ion Torrent library kits. Additional adapter sequences can be added to the list through Admin interface's Configure tab in the Torrent Browser.
Beginning with the 4.2 release, the
--trim-adapter
parameter supports multiple comma-separated adapter sequences.
Removal of lower-quality 3' ends with low quality scores
The distribution of quality scores within Ion Torrent reads is such that the highest quality calls tend to occur at the start of the read where signal is strongest and phase errors are smallest in magnitude. For reads that run into low-quality bases before reaching the end of the template, it is helpful to trim away the lower-quality calls at the 3' end. The quality trimming is performed using the per-base quality scores (see Technical Note - Quality Score ).
The approach is to scan along the bases of the read computing a moving average in a fixed-length window, and to set the read trim point to just before the earliest (5'-most) base at which the moving average of the per-base quality scores drops below a fixed threshold. The window size is set to 30 bases, and the threshold below which the trimming occurs is a quality score of 15.
Record of trimming information
Custom tags are written out in the BAM file for every read to record adapter-trimming information.
- The ZG tag specifies, in zero-based coordinates, the flow during which the first base of the adapter was incorporated.
- The ZA tag specifies the number of library insert bases, where the library insert is defined as the sequence after the key and barcode adapter, and before the 3 adapter.
- The ZB tag specifies the number of overlapping adapter bases that are found.
-
The ZC tag is a vector of the following 4 values:
- Field 1: The z ero-based flow during which the first base of the adapter was incorporated (same as ZG)
- Field 2: The zero-based flow corresponding to the last insert base
- Field 3: Length of last insert HP
- Field 4: Zero-based index of adapter type found
Quality-trimming information can be inferred indirectly from the adapter trimming data. For reads where 3' adapter trimming was more restrictive than quality trimming, ZA equals read length and the base position of 3' quality clipping is only known to be greater than the read length. When the read length is less than the ZA tag or the adapter is not found, the read length corresponds to the base position of 3' quality clipping.
Modify read-trimming options
All read trimming is performed within the BaseCaller application, which runs on the Torrent Server for both Ion PGM and Ion Proton runs. The output from the signal processing stage of the pipeline is required as input, so for both Ion PGM and Ion Proton instruments, a run can be re-analyzed from the Base Calling stage with different read trimming options. Note that this requires that the BaseCalling input data still be present on the Torrent Server the schedule on which the BaseCalling Input data are automatically deleted can be controlled in the Data Management section of the Services tab of Torrent Browser.
If non-default trimming options are desired, they can be specified in the Command Line Args section of the re-analysis screen. The following options are available:
| Option |
Value type
|
Description, [default] |
|---|---|---|
| -d, --disable-all-filters |
on/off
|
Disable all filtering and trimming. Overrides other options. [off] |
| --trim-adapter | String | Adapter sequence to trim. Usually specified via the run planning interface in Torrent Browser. Can be altered in the Analysis Options section of the re-analysis screen, and can be overridden by specifying in the Command Line Args section of the re-analysis screen.If not specified, default is Ion P1B: [ATCACCGACTGCCCATAGAGAGGCTGAGAC] |
| --trim-adapter-cutoff | Float |
Cutoff for adapter trimming. 0=off [16] |
| --trim-adapter-min-match | Integer |
Minimum adapter bases in the read required for trimming. Matches to the 3 adapter shorter than this length will be ignored. [6] |
| --trim-qual-window-size | Integer |
Window size for quality trimming. [30] |
| --trim-qual-cutoff | Float |
Cutoff for quality trimming. 100=off [15] |
Read filtering
The goal of read filtering is to remove reads that contain insufficient high quality library sequence. The fraction of reads filtered from the final output is reported in the ISP Summary.
Removal of short reads
The trimming process described above may potentially leave very few high quality bases in a read. Any read with trimmed insert length less than eight base pairs (after removal of sequencing key and any barcode adapter) is removed from the final output. All such reads are counted as Low Quality for purposes of the ISP Summary.
Removal of adapter dimers
Typically a small number of reads will consist of sequencing adapters with no insert, or with only a very short insert. Any read with an insert of less than four base pairs is labeled as an adapter dimer, and is removed from the final output. The fraction of such reads is typically less than 1%, and is reported as Adapter Dimer in the ISP Summary.
Removal of reads lacking a sequencing key
The first four bases of library reads are always TCAG. The first four bases of test fragment reads are always ATCG. Any read that does not match one of these two keys is removed from the final output. All such reads are counted as Low Quality in the ISP Summary.
Removal of reads with off-scale (pinned) signal
If during any flow, a read records a signal that hits the lower- or upper-bound of the chips measurement dynamic range, that read is removed from the final output. All such reads are counted as Empty Wells in the ISP Summary.
Removal of polyclonal reads
An Ion Sphere Particle is clonal if all of its DNA fragments are copied from a single original template. All the fragments on such a bead are identical, and they respond in unison as each nucleotide is flowed in turn across the chip. Using the standard PGM flow order, about 44% of all nucleotide flows yield a positive signal from the chip. The data from the chip are normalized so that the 1-mers in the key yield approximately unit signal. 1-mers in the insert also yield a signal value of 1, 2-mers yield a signal of 2, 3-mers 3, and so on.
Data for a clonal ISP from a 260 flow PGM run are shown below. Slightly more than half of the flows cluster around zero, and the rest are positive. The positive flows cluster around integer values. This pattern is most clearly seen for the earliest flows, before phase effects cause some templates to respond out of sync with others.
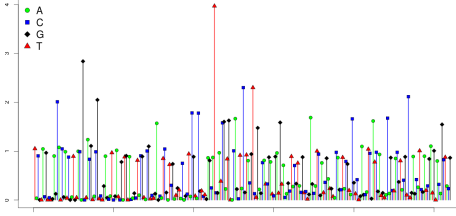
What happens if an ISP carries fragments derived from two distinct templates? The next figure shows data from such a bead. This bead harbors two distinct populations of clones. On some flows, only one of the populations responds with incorporation. Since only half the templates are incorporating, a 1-mer now yields a signal value of about 0.5 instead of 1.0; a 2-mer yields a signal of 1.0, instead of 2.0. One strand may incorporate a 1-mer, and the other a 2-mer, yielding a signal of 1.5. A signal of 0 is still possible, if both populations fail to incorporate. But such 0-signal flows are less frequent than in the clonal case, accounting for only about 19% of flows. In summary, polyclonal beads exhibit more non-zero flows, and the signals no longer cluster mainly around integers.
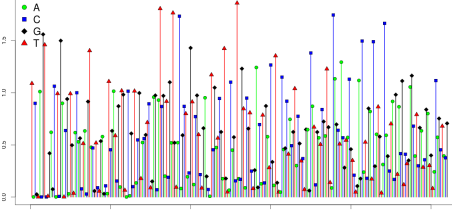
It is also possible for emPCR to generate beads carrying fragments derived from a large number of library templates. Such beads are referred to as super-mixed . The signal from a super-mixed bead is shown in the next figure. Now almost all flows have non-zero signal, and the signal does not cluster around integer values at all.
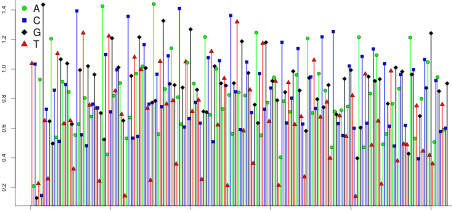
These kinds of patterns can be used to identify polyclonal beads in software. Torrent Suite does this by computing two scores for each well.
The first score is simply the percentage of flows that return a non-zero signal. Because of the possibility of noise in the data, only flows with a signal value greater than 0.25 are counted as positive. This score is computed for each bead for flows 12 through 72, and is referred to as percent positive flows ( PPF ) .
The second score captures the degree to which the signal clusters around integer values. For each flow, the difference between the signal and the nearest integer value is computed. This distance is squared, and the squared values are summed over flows 12 through 72. This scoreis low if the signal clusters around integer values, and higher otherwise. This score is referred to as the sum of squares ( SSQ ) .
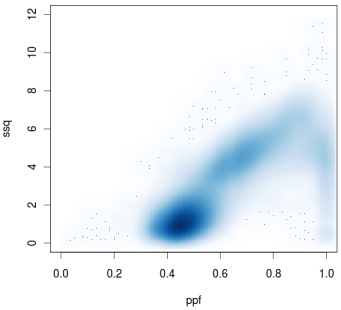
The next figure shows a scatter plot of PPF vs. SSQ for a sample of 200,000 beads from a PGM run. Three distinct populations are evident in this plot. The high density group below and to the left of the other groups has the lowest values for PPF and SSQ. Analysis of a large number of PGM runs shows that beads from this cluster are typically clonal, and map well to identifiable reference sequence. The more diffuse group at the far right has the highest PPF and SSQ scores. This group includes super-mixed beads, and other beads that fail to produce high quality sequence. The third group has intermediate PPF and high SSQ, and lies between the other two groups. This group is typically composed of mixed-template beads that fail to map to identifiable reference sequence. In this PGM run, about 74% of beads fall in the clonal cluster, 21% in the mixed cluster, and 6% in the super-mixed cluster.
The fraction of beads determined to be polyclonal is recorded in the ISP Summary.
Modify read-filtering options
Some of the read-filtering methods are implemented in the Analysis application, which performs the signal processing operation that goes from the signal traces in the DAT files (also known as the Signal Processing Input) to the estimates of incorporation for every well and flow in the wells file. The rest of the read-filtering is applied in the BaseCaller application that starts with the wells file (also known as the BaseCalling Input).
On Ion PGM instruments, both of these applications run on the Torrent Server, and the input for both stages is retained on a schedule controlled from the Data Management section of the Services tab in Torrent Browser. As a result, with Ion PGM data it is possible to reanalyze a run from the start with different read filtering options.
On an Ion Proton instrument , the Signal Processing Input for the full chip is analyzed on the instrument. Upon successful completion of analysis of the run, the Signal Processing Input is automatically deleted from the instrument. So it is not possible to re-do the full-chip Signal Processing with different read-filtering options on Ion Proton data. O nly the from-BaseCalling part of the pipeline can be repeated with different options. In cases where exploration of the read filters in the Signal Processing is desired, an alternative option is to use Ion Proton thumbnail data the Signal Processing data for the thumbnail is transferred to the Torrent Server, so the thumbnail can be reanalyzed from Signal Processing to test different options.
The following options are available to modify read filtering implemented in the BaseCaller application:
| BaseCaller option | Value | Description, [default] |
|---|---|---|
| --min-read-length | Integer |
Reads shorter than this are omitted from output. This value is inclusive of the sequencing key (which is usually TCAG), so the minimum insert length after exclusion of the sequencing key is usually 4 less than this value. [8] |
| --trim-min-read-len | Integer |
If 3 trimming reduces a read length to something shorter than this value, the read is filtered out. [8] |
The following options are available to modify read filtering implemented in the Analysis application:
| Analysis option | Value | Description, [default] |
|---|---|---|
| --clonal-filter-bkgmodel | on/off |
Controls whether or not polyclonal filtering is applied during Signal Processing. [on] |
| --mixed-first-flow | Integer |
The first flow used for evaluation of the PPF and SSQ values used for polyclonal filtering. [12] |
| --mixed-last-flow | Integer |
The last flow used for evaluation of the PPF and SSQ values used for polyclonal filtering. [72] |
Modifying read-filtering options for Ion Proton full-chip analysis (unsupported)
As explained above, there is only one opportunity to perform the Signal Processing analysis for Ion Proton full-chip runs, as the required input data is automatically deleted once successfully processed. Any attempt to change the defaults is inherently risky. If something goes wrong due to non-default settings, there is no opportunity to reanalyze and the resulting data may be compromised or lost entirely. Usage of non-default options for Ion Proton full-chip Signal Processing is unsupported and is at the users own risk. It is recommended that users test settings carefully on Ion Proton thumbnails first and proceed with caution.
The default analysis options for all chip types are accessible via the Admin Interface in the Configure tab of Torrent Browser. After logging in to the Admin Interface, click on Analysis Args , which opens a list of chip-specific defaults. Click on default_P1.1.17 to open the P1 default settings. At this point, the options described above can be set in the entries Default Analysis args and Default Basecaller args for full-chip runs, and in the entries Default Thumbnail Analysis args and Default Thumbnail Basecaller args for thumbnail runs. Take great care to avoid typos. Incorrect settings at this point may lead to loss of full-chip runs. When complete, click on the Save button in the lower-right corner.
Note that when your Torrent Suite Software is updated, these chip-specific defaults are reset to defaults and any user-applied changes will be lost. So it is recommended that details of any user customizations be saved outside of Torrent Suite Software, and after an update, any re-application of custom settings should be tested to make sure the previous customizations still work with the updated software.
