
Torrent Browser Analysis Report Guide
Torrent Suite Software space on Ion Community
Torrent Variant Caller Parameters
This page describes Torrent Variant Caller (TVC) parameters.
A note about parameter customizations
In general, you can safely customize parameters for SNP calling. For indel calling, changes to the parameters tend to have a significant effect in the number of indels called. With indels, the tradeoff between sensitivity and specificity becomes too large.
The first group of parameters are intended for general use.
Main settings
The first five parameters support different thresholds for SNP, indel, and hotspot variants. The others use the same thresholds for all variant types.
|
Parameter |
Comments |
|---|---|
|
Minimum allele frequency*
|
Minimum observed allele frequency required for a non-reference variant call. Lowering this value improves sensitivity and decreases specificity (and increases the ratio of false positives to true positives).
Allowed values
: Floats 0.0 - 1.0
|
|
Minimum quality*
|
Do not call variants if the phred-scaled call quality is below this value. Lowering this value improves sensitivity and decreases specificity.
Allowed values
: Integers >= 0
|
|
Minimum coverage*
|
Do not call variants if the total coverage on both strands is below this value. For germ line workflows, lowering coverage improves sensitivity. Lowering this value is dangerous for homopolymer indels this decreases specificity drastically.
Allowed values
: Integers >= 0
|
|
Minimum coverage on either strand*
|
Do not call variants if coverage on either strand is below this value. For indel calling, reducing this value improves sensitivity but at a high cost of specificity.
Allowed values
: Integers >= 0
|
|
Maximum strand bias*
|
Do not call variants if the proportion of variant alleles comes overwhelmingly from one strand.
Allowed values
: Floats 0.5 - 1.0
Increasing strand bias increases sensitivity. SNP calling tolerates this adjustment better than indel calling. |
|
Minimum relative read quality*
|
Do not call variants if Relative Read Quality is below this threshold. A p hred-scaled minimum average evidence per read or no-call.
Allowed values:
Floats >= 0
Impact of changing this value: Lowering this value improves sensitivity and decreases specificity. |
|
Maximum common signal shift*
|
Do not call variants if Common Signal Shift exceeds this threshold. If the predictions are distorted to fit the data more than this distance (relative to the size of the variant), filter this candidate position out.
Allowed values: Floats >= 0 Recommended: 0.3 = 30% of variant change size |
|
Maximum reference/variant signal shift (insertions)*
|
Do not call insertions if Reference or Variant Signal Shift exceeds this threshold.
Filter observed clusters that deviate from predictions by more than this amount (relative to the size of the variant).
Allowed values:
Floats >= 0
|
Maximum reference/variant signal shift (deletions)*
filter_deletion_predictions
|
Do not call deletions if Reference or Variant Signal Shift exceeds this threshold.
Filter observed clusters that deviate from predictions by more than this amount (relative to the size of the variant).
Allowed values:
Floats >= 0
|
| *Override parameters that can also be used to customize hotspot calling. See note below. | |
Note : Torrent Variant Caller v4.6 allows particular hotspots to be customized with overrides that trigger filtering. Once TVC completes, you can rerun the following parameters to override the original hotspot calls. These limits are applied per variant, not per position. Use a 6-tab delimited fields format such as: chr1 43814978 43814979 . REF=G;OBS=A;strand_bias=1; NONE. The Override Parameters are flagged with an asterisk * in the table above.
Advanced settings
These parameters allow additional customization of the variant calling algorithm but are intended for advanced users only.
Torrent Variant Caller advanced parameters
|
Parameter |
Comments |
|---|---|
|
hp_max_length |
Maximum homopolymer length for calling indels.
Allowed values
: Integers >= 1
|
|
downsample_to_coverage |
Reduce coverage in over-sampled locations to this value.
Allowed values:
Integers
>= 1
|
|
outlier_probability |
Prior probability that a read comes from some other distribution. Lower numbers reduce the influence of outlier observations. Higher numbers increase the influence of outliers.Empirical adjustment indicates that increasing the influence of outliers leads to more false-positives and slightly more true positives, but at a poor tradeoff.
Allowed values:
Floats 0.0 - 1.0
|
|
do_snp_realignment |
Realign reads in the vicinity of SNP candidates. Allowed values :
|
|
prediction_precision |
Number of pseudo-data-points suggesting our predictions match the measurements without bias. Allowed values: Floats >= 0.0 Recommended value: 1.0 Impact of changing this value: Low er ing this value increases specificity and decreases sensitivity. |
|
heavy_tailed |
How heavy the T-distribution tails are to allow for unusual spread in the data. This value represents the prior probability that a given read comes from some distribution other than the possibilities being evaluated. Lower values mean that more reads are forced to be assigned to one of the tested alleles, even at very poor data fit (fewer reads are thrown out, with the likely tradeoff of more false positive calls). Higher values mean that reads that are merely slightly noisy are thrown away, resulting in poorer sensitivity. The proportion of reads that are discarded as outliers is shown in the FXX info tag in the output VCF file. |
|
suppress_recalibration |
Ignore the base recalibration values from pipeline in TVC. (Changes the way signal is predicted.) Allowed values:
|
Long indel assembly advanced settings
These parameters control the behavior of the long indel assembler (which is a module within TVC). Again, these parameters are recommended for advanced users only.
Both the FreeBayes module and the long indel assembler generate lists of variant candidates (other modules in TVC then evaluate the candidates). The assembly module attempts to call any indel longer than 3 bp, but only reports indels that fail to be called by the FreeBase module.
|
Parameter |
Comments |
|---|---|
|
kmer_len |
Sets the length of the minimum suffix/prefix overlap (perfect match) of any two reads to be considered for assembly. Increasing this value requires longer overlaps, reducing the chances of finding matching pairs and therefore reducing the chances of calling false positives. (Increasing values make indel calls less sensitive but more specific.) Reducing the value has the opposite effect.
Allowed values
: Integers > 5
|
|
min_var_count |
Sets the number of times a variant appears in the assembled contigs in order to be considered for evaluation. Increasing this value requires more coverage of the candidate indel to be taken in consideration reducing the chances of false positive calls. (Increasing values make indel calls less sensitive but more specific.)
Allowed values:
Integers > 1
|
|
short_suffix_match |
In order for a contig to be considered for the coverage of an indel, both sides of the variant have to match perfectly the reference sequence. Increasing the size of the matching sequence sets more stringent conditions, reducing the chances of a contig to be picked as containing an indel. (Increasing values make indel calls less sensitive but more specific.)
Allowed values:
Integers > 2
|
|
min_indel_size |
Sets the minimum size of an indel (from assembled reads) to be reported. Increasing this size reduces the number (and increases the size) of the reported indels. Increasing values make indel calls less sensitive but more specific.
Allowed values:
Integers > 0
|
|
max_hp_length |
Sets the maximum length of the homopolymer to be reported. The default value has been optimized according to the physics of semiconductor sequencing. Increasing values make indel calls less sensitive but more specific.
Allowed values
: Integers > 1
|
|
min_var_freq |
Sets the minimum value of the frequency of an indel to be reported.
Allowed values
: Floats 0.0 - 1.0
Increasing this value requires a variant to be highly present in the sample in order to be called. Increasing values make indel calls less sensitive but more specific. |
|
relative_strand_bias |
Indels appearing in the sample more frequently in one strand than in the other one have an increased strand bias value. Assembled indels for which their bias value exceeds the value of this parameter are not called.
Allowed values
: Floats 0.0 - 1.0
Increasing this value makes indel calls more sensitive but less specific. |
| output_mnv |
Whether or not to include MNV variants in TVC output. Allowed values:
|
FreeBayes advanced settings
These parameters control the behavior of the FreeBayes module, which generates a list of variant candidates.
Again, these parameters are recommended for advanced users only.
|
Parameter |
Comments |
|---|---|
|
allow_indels |
Enable indels in FreeBayes hypothesis generator.
When set to 0, indels are not called.
|
|
allow_snps |
Enable
SNPs
in FreeBayes hypothesis generator. When set to 0,
SNPs
are not called.
|
|
allow_mnps |
Enable
MNP
s, including equal-length block substitutions, in the FreeBayes hypothesis generator. When set to 0,
MNP
s are not called.
|
|
allow_complex |
Enable the generation of block substitution variants candidate in FreeBayes hypothesis generator. When set to 0,
block substitution
variants
are not called.
Notes about setting allow_complex to 1:
|
|
min_mapping_qv |
Minimum mapping QV value required for reads to be allowed into the pileup. If a read has a mapping QV lower than this value, filter the position out.
Allowed values:
Integers >= 0
Impact of changing this value: Increasing this value decreases sensitivity and improves specificity. |
|
read_mismatch_limi t |
The number of mismatches allowed. If a read has more mismatches than this value, filter the read out.
Allowed values
: Integers
>= 0
|
|
read_max_mismatch_fraction |
Maximum fraction of mismatches allowed in the length of read. Filters out potentially mis-mapped reads. Allowed values: Floats 0.0 - 1.0 Recommended value : 1.0 Decreasing this value decreases sensitivity and improves specificity (fewer but more accurate reads). |
|
gen_min_alt_allele_freq |
An early-on filter for allele frequency. Filter out variant candidates that do not have at least this frequency in the pileup.
Allowed values:
Floats 0.0 - 1.0
|
|
gen_min_indel_alt_allele_freq |
An early-on filter for allele frequency for indel callings. Filter out indel candidates that do not have at least this frequency in the pileup.
Allowed values
: Floats 0.0 - 1.0
|
|
gen_min_coverage |
An early-on filter for minimum coverage. Filter out variant candidates that do not have at least this depth of coverage.
Allowed values
: Integers
>= 0
|
Advanced Settings
At the bottom of the variantCaller configuration page are two Advanced Settings text entry boxes.
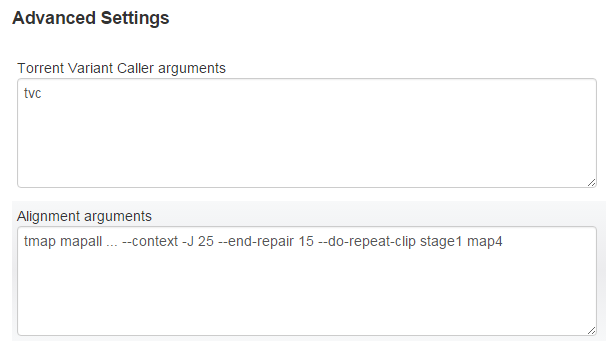
|
Parameter |
Comments |
|---|---|
|
tvc |
Torrent Variant Caller arguments |
| tmap mapall | Overrides alignment arguments if arguments entered on rerun of TVC plugin are different from arguments set in the original run. If this setting is not change, TVC does not rerun alignment arguments. |
See
 Torrent Browser Analysis Report Guide
Run Report Metrics
Run Metrics Overview
Run Report Metrics Before Alignment
Run Report Metrics on Aligned Reads
Barcode Reports
Test Fragment Report
Report Information
Output Files
Plugin Summary
Assembler SPAdes Plugin
Coverage Analysis Plugin
Torrent Browser Analysis Report Guide
Run Report Metrics
Run Metrics Overview
Run Report Metrics Before Alignment
Run Report Metrics on Aligned Reads
Barcode Reports
Test Fragment Report
Report Information
Output Files
Plugin Summary
Assembler SPAdes Plugin
Coverage Analysis Plugin
ERCC Analysis Plugin
FileExporter Plugin
FilterDuplicates Plugin
IonReporterUploader Plugin
The Ion Reporter™ Software Integration Guide
Run RecognitION Plugin
SampleID Plugin
TorrentSuiteCloud Plugin
Torrent Variant Caller Plugin
Torrent Variant Caller Parameters
Example Torrent Variant Caller Parameter File
Torrent Variant Caller Output
The Command-Line Torrent Variant Caller
Ion Reporter™ Software Features Related to Variant Calling
Integration with TaqMan® and PCR