
Torrent Browser Analysis Report Guide
Torrent Suite Software space on Ion Community
Torrent Variant Caller Output
Introduction
This page describes the Torrent Variant Caller (TVC) output, which includes the following reports and sections.
-
Run report plugin summary
-
Variant Caller Report
-
Variant Caller Report s ummary section
-
Variant Caller Report Variant Calls table
Run report plugin summary
The run report contains a short summary of plugin output. These summaries appear below the run report metrics and above the Output Files section.
To jump to the plugin results:
-
Click the Plugin Summary jump near the top of the run report:
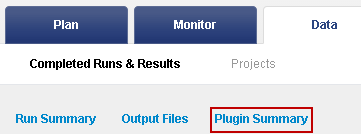
-
Click the link to go to the TVC results summary:

-
The browser jumps to the variantCaller summary.
The variantCaller summary area is slightly different for barcoded and non-barcoded runs. In both cases, the summary section includes the following:
-
Information about the analysis type, targeted regions and hotspot files, and variantCaller parameter settings.
-
The total number of variants called.
- The variantCaller.html link to the results page.
-
Download links:
- The zipped VCF file of variant calls.
- The Zipped VCF index file (required for IGV).
- The results in a tab-separated file.
Barcoded variantCaller summary area
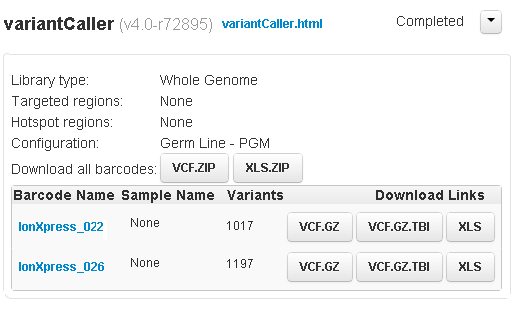
For a barcoded run:
- When the run contains multiple barcodes, the variantCaller.html link opens a listing of the barcodes.
-
Links to a separate results page for each barcode.
- A link to download all results in one zipped file.
Non-barcoded variantCaller summary area
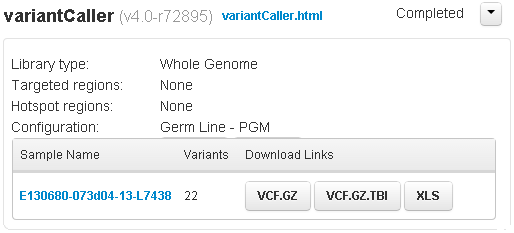
For a non-barcoded run, the sample name is listed. This link and the variantCaller.html link open the same results page.
View plugin log file
To view the log file for a plugin run, in the plugin summary area, click the menu arrow next to the plugin status:
Delete plugin results
To delete the results for a plugin run, in the plugin summary area, click the menu arrow next to the plugin status:
This action removes the plugin's output files from the file system and removes the Torrent Browser plugin results page.
Variant Caller Report
The open the plugin report, click the
variantCaller.html
link in the run report Plugin Summary area:

The plugin report begins with a listing of information and download links, as described in the following tables:
Information fields
|
Field |
Description |
|---|---|
|
Run |
The run report name |
| Barcode | Barcode name (only for barcdoed runs) |
|
Sample Name |
Name of the sample |
|
Reference |
Name of the reference used in the analysis |
|
Library Type |
Library type |
|
Variant Caller Version |
Version of the plugin |
Buttons for downloads and other actions
|
Field |
Button |
Description |
|---|---|---|
|
Targeted regions |
BED |
Downloads the input targeted regions BED file (if any). |
|
Hotspot regions |
BED |
Downloads the input hotspots BED file (if any). |
|
Parameters Settings |
Parameters File |
Downloads a JSON text file of the TVC parameter values used on this run. Note: You can edit this file and later upload it to set your custom parameters in subsequent runs. For an example of this type of file, see Example Torrent Variant Caller Parameter File . |
|
Mapped Reads |
BAM, BAI |
Downloads t he BAM file (and its index) of mapped reads. This file is input to TVC. |
|
Variant Calls |
VCF.GZ,
|
Downloads files of the variants calls:
VCF.GZ,
VCF.GZ.TBI: Zipped VCF file and its tabix index file
|
|
Open Variants Calls in IGV |
IGV |
Link to open the results variants in the Integrated Genomic Browser (IGV). |
|
Deprecated Features |
Classic |
Opens the plugin results page in the previous format. |
| Ion Community | Torrent Variant Caller documentation | Opens to the Torrent Variant Caller documentation page on the Ion Community (login is required). |
VCF File
The VCF files contains all the information used by the other summary tables and all the variant calls. In its header it also contain a QC metric useful for FFPE samples:
Deamination Metric
The deamination metric denotes the quality of an FFPE sample. The deamination score is the fraction of C>T plus G>A type even counts (at read level) within all substitution event counts. Currently, the following basic requirements apply for a substitution event to be counted:
1. The substitution position needs to be within the amplicon insert region for the amplicon that a read is assigned to.
2.The substitution position needs to have >30 coverage on both sides.
3. The ratio of a particular type of substitution at a given position needs to be >0.001 and <0.15 on both strands.
Variant Calls by Allele table
The following list summarizes the features of the Variant Calls table:
-
Each position is a link to open the variant in IGV. In some browsers, you save the
igv.jnlpfile to your local system, and then click onigv.jnlpto open the IGV browser. -
You can export selected variants to a table file or to the Thermo Fisher PCR and Sanger Sequencing Por TaqMan Assay Design web sites. See Export to File , Export to Pre-Designed Primers for PCR and Sanger Sequencing , Export to TaqMan Assay Design .
-
Click on a column header to order the table by the contents of that column.
- For candidates that are filtered out, the filtering reason is highlighted in the table. For example:
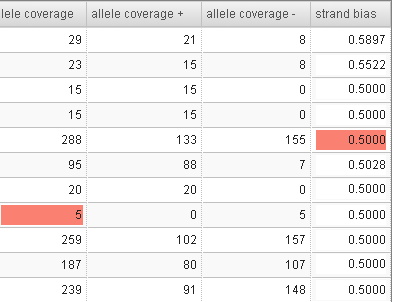
The main columns are described in the following table. Use the View tabs on the right of the table to change the display of the columns on the right:

|
Column |
Description |
|
Position |
The chromosome (or contig) name in the reference genome, and t he one-based position in the reference genome. |
|
Ref |
The reference base(s). |
|
Variant |
Variant allele base(s). |
|
Var Freq |
Frequency of the variant allele. |
|
Quality |
Phred-scored quality field.Larger values mean more certainty in the call. Typically very large for reads strongly distinguishing variants (SNPs) with good depth; that is, under the model assumed, evidence is overwhelming for the variant or for the reference. Marginal values in this field can mean either the reads do not distinguish the variant well or there is insufficient depth to resolve, or the observed allele frequency is near the cutoff.Filters to compensate for the cases in which the model assumptions are not true are found in the INFO tags. Computed by posterior probability that the sample variant allele frequency is greater than the min-allele-frequency specified for the variant type (if a variant), or posterior probability that the variant allele frequency is below this threshold (if a reference call).Posterior probability computed conditional on the reads observed, includes sampling variability. |
View Allele Annotations
These columns are displayed in the run report in the View Allele Annotations tab:
| Column | Description |
|---|---|
| Variant Type |
SNP Single nucleotide polymorphism IND Insertion DEL Deletion MNP Multiple nucleotide polymorphism COMPLEX Complex block substitution |
| Allele Source |
Hotspot if called only because of its entry in a hotspots file Novel all others |
| Allele Name | The Allele name as given in the target regions file |
| Gene ID | The Gene ID as given in the target regions file |
| Region Name | The r egion name as given in the target regions file |
View Coverage Metrics
These columns are displayed in the run report in the View Coverage Metrics tab:
| Column | Description |
|---|---|
| Coverage | Total coverage at this position, after downsampling |
| Coverage + | Total coverage on the forward strand, after downsampling |
| Coverage - | Total coverage on the reverse strand, after downsampling |
| Allele Cov | The number of reads that contain this allele, after downsampling |
| Allele Cov + | Allele coverage on the forward strand, after downsampling |
| Allele Cov - | Allele coverage on the reverse strand, after downsampling |
| Strand bias |
Discrepancy between allele frequencies on the forward and reverse strands |
View Quality Metrics
These columns are displayed in the run report in the View Quality Metrics tab. Associated filtering codes are given in brackets.
| Column | Description |
|---|---|
| Common Signal Shift |
Distance between predicted and observed signal at the allele locus. [RBI] |
| Reference Signal Shift |
Distance between predicted and observed signal in the reference allele. [REFB] |
| VariantSignal Shift |
Distance between predicted and observed signal in the variantallele. [VARB] |
| Relative Read Quality |
Phred-scaled mean log-likelihood difference between the prediction under reference and under the variant hypothesis. [MLLD] |
| HP Length | Homopolymer length. |
| Context Error + |
Probability of sequence-specific error on the forward strand (reported only for deletion variants). |
| Context Error - |
Probability of sequence-specific error on the reverse strand (reported only for deletion variants). |
| Context Strand Bias |
Basespace strand bias ( reported only for deletion variants ). |
Filtering codes
This table lists TVC filtering codes, which describe why a candidate is filtered out and not considered a variant.
|
Filtering reason |
Description |
|---|---|
|
COVERAGE |
Corresponds to the various counting VCF tags on each strand for alleles: FDP, FSAF, FSAR, FSRF, FSRR. Set by the TVC parameter
|
|
FAIL |
The filtering is based on the quality score (probability that the variant at the first location has an allele-frequency above the specified location, or for a reference call, that we can exclude the variant having an allele-frequency above the specified frequency), controlled by
Note
:
|
|
HPLEN |
Corresponds to the VCF tag HRUN. It is set by TVC parameter
|
|
PREDICTIONSHIFT |
Corresponds to the VCF tag RBI, which measures the amount by which the predictions had to be distorted to match the measurements. A value such as 0.3=30% of the difference the variant would make to the read (e.g., a shift of 0.3 for a 1 base deletion, a shift of 0.6 for a 2-base deletion). Set by TVC parameter
|
|
REFERENCE |
We generate a candidate variant at this location, but after considering the evidence, generate a 0/0 genotype call at the location. Because this clutters the VCF file with entries at locations that are not variants, there is a flag
|
|
SSE |
Corresponds to the VCF tags SSEN and SSEP, and means that at this location we predict a potential error due to the local sequence context, on either the positive or negative strand.Set by the two TVC internal filter controls
|
|
STDBIAS |
Corresponds to the VCF tag STB.Filters out variants if the stand bias for the variant is too large. Set by TVC parameter
|
|
STRINGENCY |
Corresponds to the VCF tag MLLD, which is mean-log-likelihood difference per read for the variant. This is approximately phred-scaled, so that MLLD=10 = 10% error rate per read, MLLD=20 = 1% error rate per read. Variants are filtered by the TVC parameter
|
Export to file
This option exports your variant calls to a tab-separated file.
The exported file is named
subtable.xls
and has the same columns as the Variant Calls table (including columns for all three display options: View Allele Annotations,
View Coverage Metrics, and View Quality Metrics).
Click the left column checkboxes to select your variants, then click the Export Selected button:
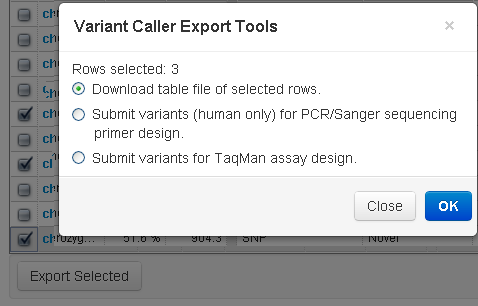
Export to Pre-Designed Primers for PCR and Sanger Sequencing site
With this export option, you select variants to search for pre-designed primers for PCR and Sanger sequencing.
To initiate a search, use the checkboxes column (on the left) to select variants from the TVC plugin report and click on the Export Selected button. A pop-up window allows you to submit variants for the PCR and Sanger sequencing pre-designed primers site:
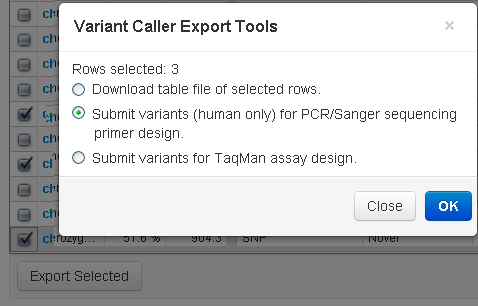
A new browser window opens to this site: Pre-Designed Primers for PCR and Sanger Sequencing . You must be connected to the internet to access this feature.
TaqMan Assay Search
After running the TVC plugin, you can select variants from a report table and submit a search against the TaqMan Assay Search webpage. When the search is submitted, you can choose which TaqMan Assay database to search. A new browser window appears with the search results. If TaqMan assays are available for the selected variant, you can immediately order the assay directly from the landing page. You must be connected to the internet to access this feature.
To initiate a search, use the checkboxes column (on the left) to select variants from the TVC plugin report and click the Export Selected button. A pop-up window allows you to submit variants for TaqMan assay design.
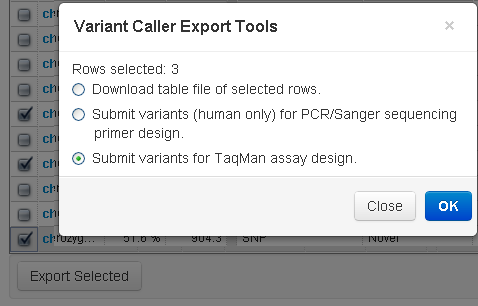
You are then presented with a list of verification assays that you can order from Thermo Fisher.
See
 Torrent Browser Analysis Report Guide
Run Report Metrics
Run Metrics Overview
Run Report Metrics Before Alignment
Run Report Metrics on Aligned Reads
Barcode Reports
Test Fragment Report
Report Information
Output Files
Plugin Summary
Assembler SPAdes Plugin
Coverage Analysis Plugin
Torrent Browser Analysis Report Guide
Run Report Metrics
Run Metrics Overview
Run Report Metrics Before Alignment
Run Report Metrics on Aligned Reads
Barcode Reports
Test Fragment Report
Report Information
Output Files
Plugin Summary
Assembler SPAdes Plugin
Coverage Analysis Plugin
ERCC Analysis Plugin
FileExporter Plugin
FilterDuplicates Plugin
IonReporterUploader Plugin
The Ion Reporter™ Software Integration Guide
Run RecognitION Plugin
SampleID Plugin
TorrentSuiteCloud Plugin
Torrent Variant Caller Plugin
Torrent Variant Caller Parameters
Example Torrent Variant Caller Parameter File
Torrent Variant Caller Output
The Command-Line Torrent Variant Caller
Ion Reporter™ Software Features Related to Variant Calling
Integration with TaqMan® and PCR