
Torrent Browser Analysis Report Guide
Torrent Suite Software space on Ion Community
Output Files
These links permit you to directly download the data and report files.Some files are compressed, using the
.zip
format, to provide data integrity and to reduce download time.
Click on a file type button to save the file to your local computer.Most outputfiles can be loaded into third-party viewers (such as IGV) for visualization. The barcode row only appears for runs on barcoded data.
Files in the barcode row are zips of one file per active barcode. To download only BAM and BAI files for a single barcode, go to the barcode section at the top of the run report (see Barcode Reports ).
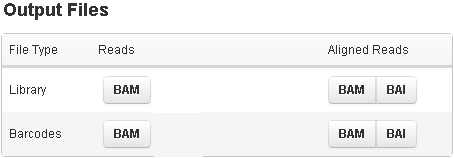
| Column | Description |
|---|---|
| Reads | Files with unaligned reads (before alignment) |
| Aligned Reads | Files with aligned reads |
| File type | Reads | Aligned reads |
|---|---|---|
| BAM |
Unaligned reads in BAM format. In this release, the BAM file contains some flow space information. |
Aligned reads sorted by reference location. See Run Metrics Overview for a description of the alignment data included in this BAM file. |
| BAI | BAM index file |
The BAM format
Binary Sequence Alignment/Map (BAM), is a compressed, binary form of the SAM format. BAM files can be indexed, using the BAM Index file, for quick access to sequence alignment data. See http://samtools.sourceforge.net for more a more detailed description of the SAM/BAM file format. Many tools are available for working with SAM files.
Deprecated file formats
The following file formats are deprecated and not produced by the default analysis pipeline. The FileExporter Plugin optionally generates these files.
.
| File type | Description |
|---|---|
| SFF |
Compressed (.zip) Standard Flowgram Format (SFF) -formatted file that contains "flow space" data.
The data are organized on a per flow basis, and contain information about nucleotide flows that both did and did not result in base incorporation. (See Technical Note - Filtering and Trimming .) Note: The SFF file format is deprecated and not produced by the default pipeline. |
| FASTQ |
Compressed (.zip)FASTQ-formatted file containing data organized in a per-base basis, including quality scores. The reads contained in the file are unaligned reads. (See Technical Note - Filtering and Trimming .) Note: The FASTQ file format is deprecated and not produced by the default pipeline. |
Rename your output files
See the FileExporter Plugin supports renaming your putput files. (This plugin also optionally creates SFF or FASTQ formats, or zips your output.)
 Torrent Browser Analysis Report Guide
Run Report Metrics
Run Metrics Overview
Run Report Metrics Before Alignment
Run Report Metrics on Aligned Reads
Torrent Browser Analysis Report Guide
Run Report Metrics
Run Metrics Overview
Run Report Metrics Before Alignment
Run Report Metrics on Aligned Reads
Barcode Reports
Test Fragment Report
Report Information
Output Files
Plugin Summary
Assembler SPAdes Plugin
Coverage Analysis Plugin
ERCC Analysis Plugin
FileExporter Plugin
FilterDuplicates Plugin
IonReporterUploader Plugin
See
The Ion Reporter™ Software Integration Guide
Run RecognitION Plugin
SampleID Plugin
TorrentSuiteCloud Plugin
Torrent Variant Caller Plugin
Torrent Variant Caller Parameters
Example Torrent Variant Caller Parameter File
Torrent Variant Caller Output
The Command-Line Torrent Variant Caller
Ion Reporter™ Software Features Related to Variant Calling
Integration with TaqMan® and PCR