
Torrent Browser User Interface Guide
Torrent Suite Software space on Ion Community
Reanalyze a run
To begin a sequencing run, see Templates , Planned Runs , and Plan by Sample Set .
When you reanalyze, any setting changes you make affect only the current run (when you click the Start Analysis button). If you instead click Edit , your changes are saved for each subsequent reanalysis. See also Edit run metadata .
Start your reanalysis
You select the Reanalyze option either from the Completed Runs And Reports table or from a run report:
-
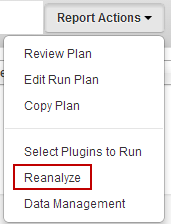
In the run report of a completed run, click the
Report Actions > Reanalyze
in the report header:
-
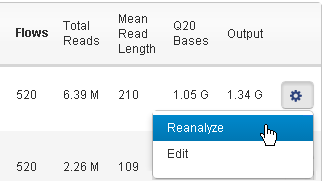
In the
Data > Completed Runs & Results
tab table view, click the
Reanalyze
option in the gear menu on the right of the run entry:
Follow these steps to reanalyze your run:
-
Use one of the Reanalyze options.
-
Enter the Report Name for the new run (required).
-
To rerun the analysis with the same settings , fill in the report name and click Start Analysis .
To rerun the analysis with changed settings, click the tabs on the left panel to display the analysis options and make your changes.
-
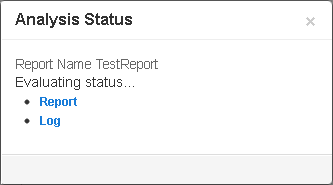
Click Start Analysis to start the run, which checks the analysis status before displaying the run confirmation message:
-
View run progress by clicking
Report
or by selecting the run name in the
Monitor
tab.
- Click Log to view run progress details in text form. Click your browser refresh button to update the log.
Settings tabs
When you reanalyze a run, you have to option to change the run's settings. Settings are organized by tab in the left panel:
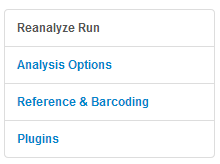
The settings for your original run are displayed when you open these tabs.
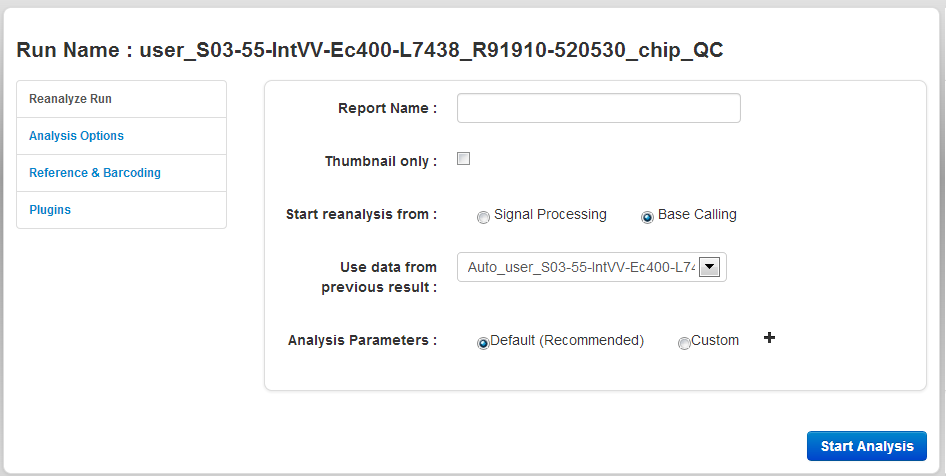
The Reanalyze Run tab
In the Reanalyze tab, you enter a name for the new run report and the reanalysis starting point:
| Setting | Description |
|---|---|
| Report Name | The name of the new run report (the result of the reanalysis). |
| Start reanalysis from |
The Analysis Pipeline proceeds through three stages: Signal Processing, Base Calling, and Alignment. Normally report generation proceeds through all three steps, but if you have already generated a report, it is possible to reanalyze the experiment and skip the earlier stages of the pipeline. For example, you may wish to change the genome that is used for Alignment. After changing the genome for the experiment on the Runs page using the Edit field, you need to reanalyze data to produce a new report using the new genome. But since there is no need to repeat the time consuming Signal Processing and Basecalling steps, you can use the output from an existing report as a starting point for Alignment, and the report will be completed much more quickly. You can restart the analysis from these points:
|
| Use data from previous report | This option applies only when starting reanalysis from Base Calling. In this case, the results from a previous report are used as input for reanalysis. |
Analysis Parameters
Click Custom to view the Analysis Parameters page:
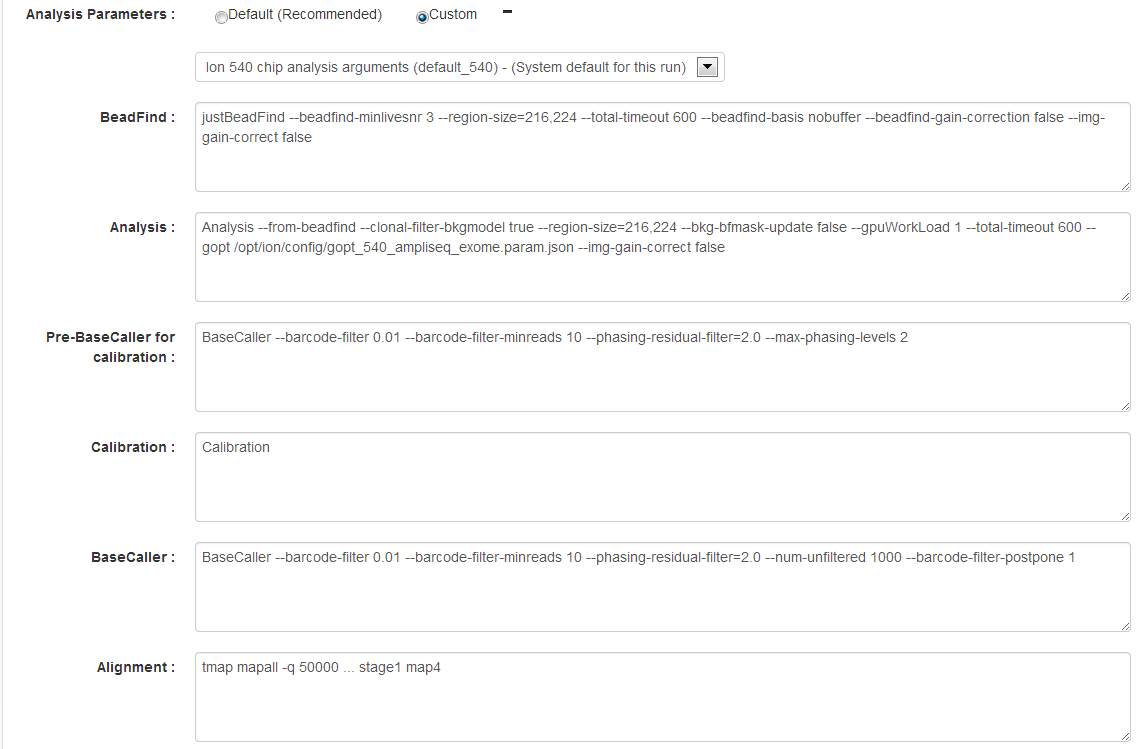
| Setting | Description |
|---|---|
|
Beadfind args |
Beadfind module command line arguments. Should not be modified unless instructed by Ion Torrent Technical Support. |
|
Analysis args |
Analysis command line arguments. Should not be modified unless instructed by Ion Torrent Technical Support. |
| Pre Basecaller args for calibration |
BaseCaller command line arguments. See
Basecaller arguments
for information on
This field is used only if the Enable Recalibration checkbox is enabled. |
| Recalibration Args |
Recalibration command line arguments.
The Recalibration
|
| Basecaller args |
BaseCaller command line arguments. See
Basecaller arguments
for information on
--barcode-mode
,
--barcode-cutoff
, and
--barcode-filter
. Other Basecaller arguments should not be modified unless instructed by Ion Torrent Technical Support.
|
| Alignment Args |
Arguments for the TMAP aligner. (Replaces the TMAP Args field that appears in previous releases.) |
See TMAP Modules for information about TMAP parameters.
BaseCaller arguments
This section describes select arguments used with the BaseCaller module.
| Argument | Type | Description |
|---|---|---|
| --barcode-cutoff |
1.0 (Float) |
Maximum distance allowed in barcode matches.A threshold that sets the stringency for barcode matches. Lower values require more exact matches when assigning reads to barcodes. Higher values allow less exact matches. Reads that have a distance greater than this value are counted as barcode no-matches. |
| --barcode-filter |
0.01 (Float) |
Barcode frequency threshold to be reported in the UI. Set to 0.0 to turn this filter off. The setting 0.0 causes all barcodes in the barcode set to be reported in the UI (including barcodes with no or very few reads). |
| --barcode-mode |
2 (Integer) |
Allowed values: 1, 2
Note : --barcode-mode 0 is no longer supported. |
| --barcode-filter-minreads |
20 (INT) |
Threshold for the minimum number of reads in a barcode group, for that group to be reported in the UI. . |
See BaseCaller Parameters for information about other, less-frequently-used parameters.
The Analysis Options tab
An example Analysis Options page is shown here:
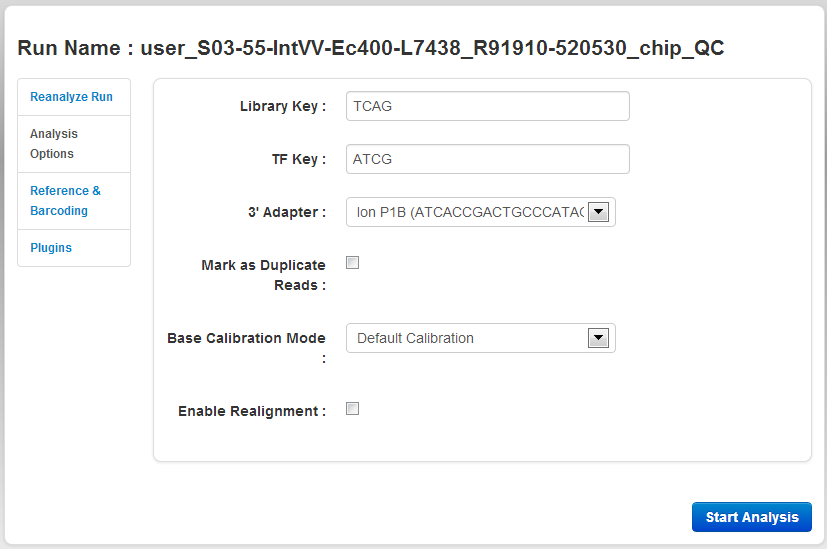
The Analysis Options tab contains these settings:
| Setting | Description |
|---|---|
|
Library Key |
Sequence used to identify library reads. Example: "TCAG". |
| TF key | Sequence used to identify test fragment reads. Example: "ATCG". |
|
TF config |
Name of an external file that specifies Test Fragment information. If a file is not specified, the default Test Fragment file is used. |
| 3' Adapter | The name and sequence of the 3' adapter. |
| Mark as Duplicate Reads | Filter out PCR duplicates. Useful when reanalyzing combined BAM files. Do not use with Ion AmpliSeq data. See About the Mark as Duplicate Reads . |
| Base Calibration Mode | Three options are available: Default Calibration, No Calibration and Enable Calibration Standard. See topic 'About the Base Calibration mode options' here: Wizard Kits Chevron . |
| Enable Realignment |
Realignment is an optional analysis step that is executed right after TMAP. This step adjusts the alignment, primarily in the CIGAR string. |
About the Mark as Duplicates Reads option
For some applications, duplicate reads coming from PCR cause problems in downstream analysis. The presence of duplicate reads may create the appearance of multiple independent reads supporting a particular interpretation, when some of the reads are in fact duplicates of each other with no additional evidence for the interpretation.
Torrent Suite Software uses an Ion-optimized approach that considers the read start and end positions by using both the 5' alignment start site and the flow in which the 3' adapter is detected. Duplicate reads are flagged in the BAM in a dedicated field. Use of the Torrent Suite Software method is recommended over other approaches which consider only the 5' alignment start site.
Marking duplicate reads is not appropriate for Ion AmpliSeq data, because many independent reads are expected to share the same 5' alignment position and 3' adapter flow as each other. Marking duplicates on an Ion AmpliSeq run risks inappropriately flagging many reads that are in fact independent of one another.
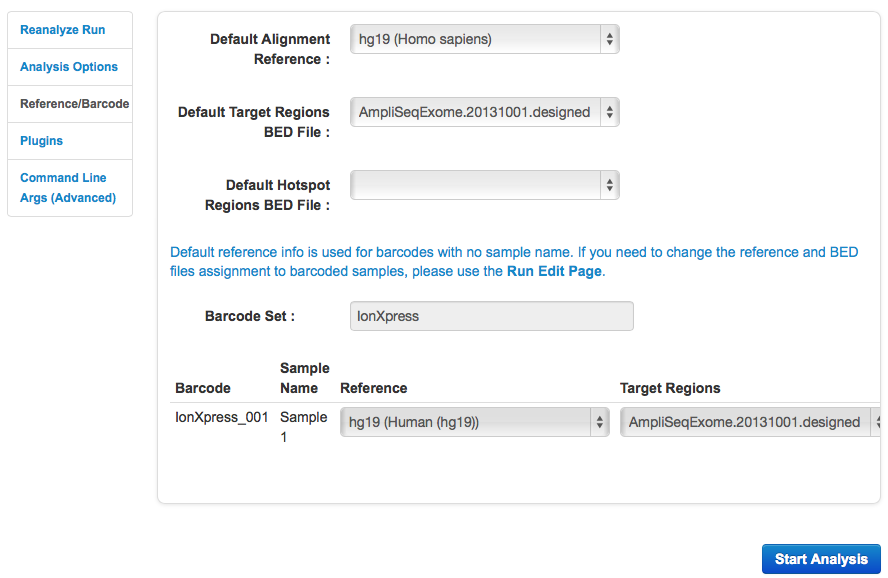
The Reference & Barcoding tab
The Reference & Barcoding tab contains these settings:
| Setting | Description |
|---|---|
| Default Alignment Reference | The genomic reference to align to. Use this menu to change the reference used for alignment in the new analysis. |
| Default Target Regions BED File | Targeted regions of interest file. Analysis is restricted only to regions listed in this file. |
| Default Hotspot Regions BED File | Hotspots file. The variant caller includes each hotspot position in its output VCF file. Variant caller filter scores are provided for each hotspot position that does not have a variant called. |
| Barcode Set | The DNA barcode set. |
See TMAP Modules for information about TMAP parameters.
 Torrent Browser User Interface Guide
The Login Page
The Plan Tab
Torrent Browser User Interface Guide
The Login Page
The Plan Tab
Templates
Planned Runs
Plan by Sample Set
Create Samples and a Sample Set
Sample Attributes
Template and Planned Run Wizard
Create Multiple Run Plans
Create a Template with Ion AmpliSeq.com Import
The Monitor Tab
The Data Tab
Completed Runs and Reports Tab
Work with Completed Runs
Reanalyze a Completed Run
BaseCaller Parameters
TMAP Parameters
The Projects Listing Page
Project Result Sets Page
 Compare Multiple Run Reports
CSV Metrics File Format
Compare Multiple Run Reports
CSV Metrics File Format